General Usage¶
XPRESSpipe can be run essentially from beginning to end as a pipeline, or as individual sub-modules. We will describe each option in more detail in each section of the documentation. The purpose of XPRESSpipe is to automate the alignment, quality control, and initial analysis of single-end (SE), paired-end (PE), and ribosome profiling data. It is intended that input data is in its own directory and that each file is a properly formatted
fastq file. However, the suffix for these files can be fq or txt as well. They can be zipped (zip or gz) or unzipped. When using intermediate sub-modules, such as align or readDistribution, input will vary and is explicatedin the --help menu for each sub-module.
Further analysis on the resulting datasets can be performed using XPRESSplot.
File Naming¶
In order for many of the XPRESSpipe functions to perform properly and for the output to be reliable after alignment (except for generation of a raw counts table), file naming conventions must be followed.
1. Download your raw sequence data and place in a folder – this folder should contain all the sequence data and nothing else.
2. If you are working with single-end data, the files must be a FASTQ-formatted file and end with the suffix
fastq, fastq.gz, fq, fq.gz, txt, txt.gz. We recommend the fastq or fastq.gz suffix.3. If you are working with paired-end data, the rules from
Step 2 apply, but must the suffix must be prefaced by the paired read group number as below:ExperimentName_Rep1_a_WT.r1.fastq.gz
ExperimentName_Rep1_a_WT.r2.fastq.gz
ExperimentName_Rep2_a_WT.r1.fastq.gz
ExperimentName_Rep2_a_WT.r2.fastq.gz
or
ExperimentName_Rep1_a_WT.read1.fastq.gz
ExperimentName_Rep1_a_WT.read2.fastq.gz
ExperimentName_Rep2_a_WT.read1.fastq.gz
ExperimentName_Rep2_a_WT.read2.fastq.gz
Data Output¶
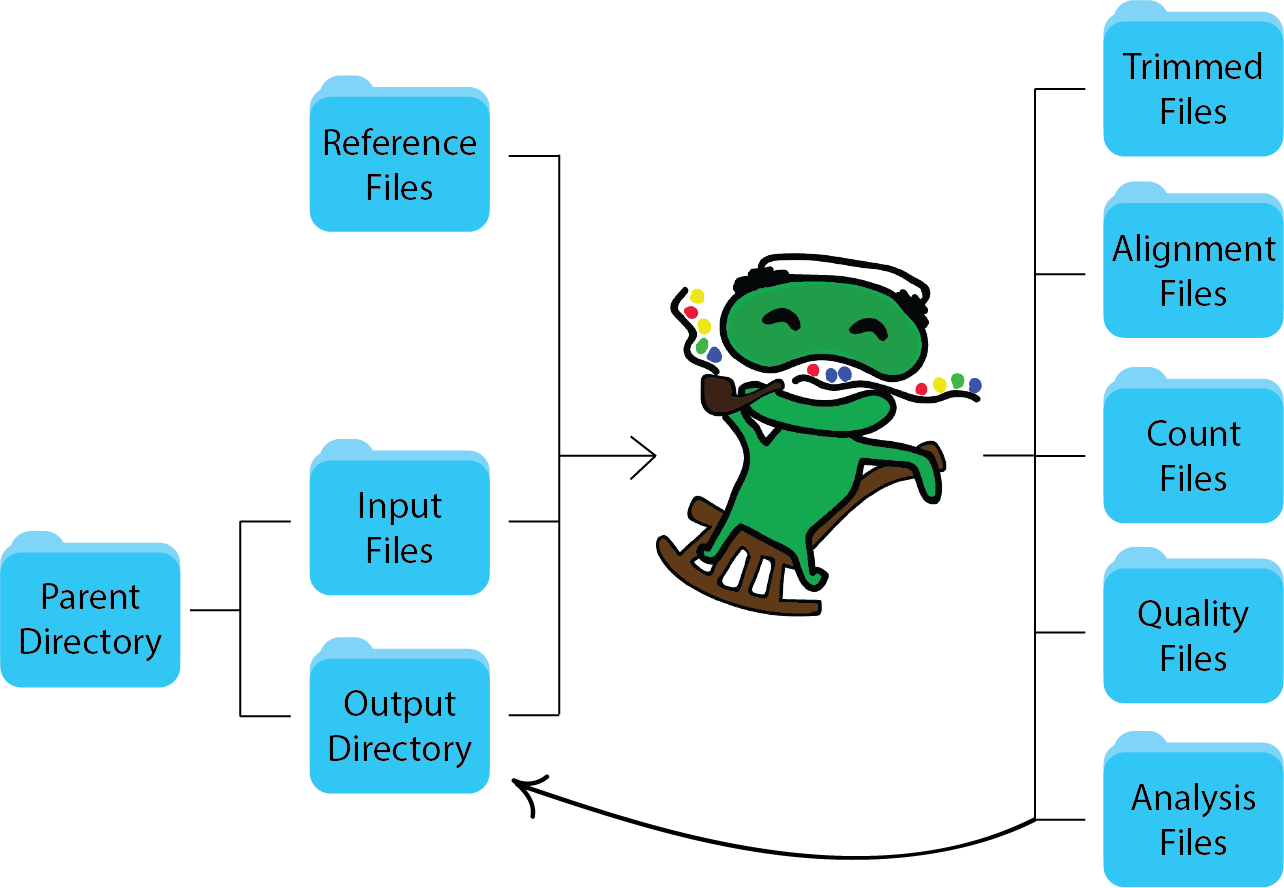
Running seRNAseq, peRNAseq, or riboseq will output all intermediate and final data files as shown in this schematic: